


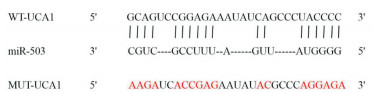
-
目前心血管疾病是世界上发病率和死亡率最高的疾病。急性心肌梗死(AMI)是心血管疾病的常见类型,已对人类健康构成巨大威胁。冠状动脉粥样硬化血管狭窄引起心肌缺血和缺氧,长期缺血甚至导致心肌细胞死亡,最终导致AMI [1-2]。然而,AMI发病机制并未完全阐明。非编码RNA(ncRNA)是一类缺乏蛋白质编码能力的RNA,最初被认为是“垃圾DNA”。近年研究[3]发现,人类基因组约98%为ncRNA,长链非编码RNA(lncRNA)和微小RNA(miRNA)是ncRNA的重要类型,参与细胞增殖、周期进展、迁移和凋亡等多种细胞过程,具有强大的基因调控功能。此外,lncRNA还可与miRNA相互作用防止目标mRNA被miRNA降解[4]。研究[5]显示,AMI病人血浆中lncRNA尿路上皮癌相关1(UCA1)表达显著降低,是急性心肌梗死潜在的诊断标志和治疗靶点。生物信息学数据库在线分析发现,UCA1与miR-503之间存在结合位点。miR-503高表达已被证实与糖尿病心肌梗死边缘区组织内血管新生能力的下降有关[6]。然而,lncRNA UCA1能否靶向miR-503在AMI中发挥作用尚未完全阐明。本研究在缺氧条件下培养H9c2心肌细胞以诱导细胞损伤,探讨UCA1在预防缺氧引起的心肌损伤中作用和可能机制。现作报道。
-
人心肌细胞株AC16购自中国科学院上海细胞库;DMEM高糖培养基、胎牛血清购自美国Hyclone公司;空载体质粒(pcDNA)、UCA1过表达质粒(pcDNA-UCA1)、miRNA抑制物阴性对照(anti-miR-NC)、miR-503抑制物(anti-miR-503)、双荧光素酶报告基因载体购自上海生工生物工程有限公司;Trizol试剂、放射免疫沉淀试验(RIPA)裂解缓冲液、二喹啉甲酸(BCA)试剂盒、核糖核酸酶A(RNase A)、兔抗Ki67抗体、兔抗激活型半胱氨酸酶3(Cleaved-caspase3)抗体、兔抗磷酸甘油醛脱氢酶(GAPDH)抗体、山羊抗兔IgG购自上海碧云天生物研究所;cDNA合成试剂盒、miRNA反转录试剂盒、SYBR green混合物试剂、Taqman Universal混合物试剂购自大连Takara生物;膜联蛋白-异硫氰酸荧光素/碘化丙啶(Annexin V-FITC/PI)凋亡检测试剂盒购自上海贝博生物科技有限公司。
-
AC16采用添加10%胎牛血清的DMEM高糖培养基置于含5% CO2、37 ℃培养箱中培养。按照1:3比例传代,每周3次。取2×105个对数期AC16细胞接种24孔板,置于缺氧培养箱中培养(5% CO2、1% O2、94% N2、37 ℃)的缺氧培养箱48 h,记为模型(Model)组。同时设置对照组不做缺氧处理。收集细胞进行后续实验。为证实lncRNA UCA1、miR-503在缺氧诱导的AC16细胞损伤中的作用,将AC16细胞分为pcDNA组、pcDNA-UCA1组、anti-miR-NC组、anti-miR-503组,即利用脂质体转染法将pcDNA、pcDNA-UCA1、anti-miR-NC、anti-miR-503分别转染AC16细胞,转染48 h检测转染效率合格后进行缺氧处理。
-
Trizol试剂提取细胞总RNA。分别使用cDNA合成试剂盒、miRNA反转录试剂盒进行逆转录反应,分别使用SYBR green混合物试剂、Taqman Universal Master Mix Ⅱ进行RT-qPCR,2-△△CT法分析lncRNA UCA1(内参为GAPDH)和miR-503(内参为U6)表达量。miR-503引物F: 5′-CCT ATT TCC CAT GAT TCC TTC ATA-3′,R:5′-GTA ATA CGG TTA TCC ACG CG-3′,扩增片段长度为159 bp;UCA1引物F:5′-TTT GCC AGC CTC AGC TTA AT-3′,R:5′-TTG TCC CCA TTT TCC ATC AT-3′,扩增片段长度为320 bp;U6引物F:5′-GTG ATC ACT CCC TGC CTG AG-3′,R:5′-GGA CTT CAC TGG ACC AGA CG-3′,扩增片段长度为201 bp;GAPDH引物F:5′-CCG CAT CTT CTT GTG CAG TG-3′,R:5′-CCC AAT ACG GCC AAA TCC GT-3′,扩增片段长度为450 bp。
-
取5×103个细胞接种96孔板,培养24 h后每孔添加10 μL的CCK-8溶液,培养箱再孵育2.5 h,酶标仪在450 nm处测定各孔吸光度(A)。细胞存活率=对照组A/实验组A×100%。
-
凋亡检测:采用结合缓冲液将LPS处理24 h的各组细胞调整为1×105/mL单细胞悬液。取100 μL细胞悬液加入到流式管,按照凋亡检测试剂盒说明书加入Annexin V-FITC和PI试剂进行染色,避光反应15 min,1 h内上机检测细胞凋亡。
周期分布检测:常规消化各组细胞,离心弃去上清液,用预冷的PBS洗涤细胞2次。用预冷的70%乙醇室温固定1 h。离心收集细胞,PBS洗涤细胞2次。向细胞中加500 μL RNase A(10 μg/mL), 室温孵育30 min。再加500 μL PI(100 μg/mL), 室温孵育30 min。流式细胞仪检测各时相细胞比例。
-
应用lncRNABase在线预测显示lncRNA UCA1与miR-503存在结合位点。基于预测结合位点合成UCA1野生型(WT)和突变型(MUT)序列,将上述序列分别克隆pmirGLO荧光素酶载体构建WT-UCA1、MUT-UCA1。将miR-503 mimics、miR-NC与WT-UCA1或MUT-UCA1分别共转染至A549细胞48 h后,双荧光素酶报告基因检测试剂盒测定相对荧光素酶活性。将pcDNA-UCA1、pcDNA分别转染AC16细胞,转染48 h RT-qPCR法检测miR-503表达水平。
-
实用RIPA裂解缓冲液提取细胞总蛋白,BCA试剂盒分析蛋白质浓度。将蛋白样品加至聚丙烯酰胺凝胶加样孔分离蛋白,并进行湿法转膜。用含5%脱脂牛奶的缓冲液封闭膜1 h,将抗Ki67抗体、抗Cleaved-caspase3抗体、抗GAPDH抗体按照1:1 000比例稀释,4 ℃孵育膜过夜。洗膜缓冲液冲洗3次,用1:1 000稀释的二抗溶液孵育膜1 h。洗膜缓冲液再次冲洗3次。将膜与化学发光显色底物孵育1 h,Image LabTM软件分析蛋白条带灰度值,目的蛋白与GAPDH灰度值比值表示目的蛋白表达水平。
-
采用t检验、方差分析和q检验。
-
相对于对照组,模型组AC16细胞中UCA1表达降低,miR-503表达升高,差异有统计学意义(P < 0.01)(见表 1)。
分组 n UCA1 miR-503 对照组 3 0.99±0.09 0.96±0.10 模型组 3 0.12±0.02 2.67±0.12 t — 16.34 18.96 P — < 0.01 < 0.01 表 1 UCA1、miR-503在缺氧处理的AC16细胞中的表达(x±s)
-
相对于pcDNA组(0.12±0.01),pcDNA-UCA1组(0.63±0.04)AC16细胞中UCA1表达显著升高(t=21.42,P < 0.01)。相对于anti-miR-NC组(2.68±0.13),anti-miR-503组(1.16±0.06)AC16细胞中miR-503表达显著降低(t=18.39,P < 0.01)。
-
相对于对照组,模型组AC16细胞存活率、Ki67蛋白表达、S期细胞比例降低,细胞凋亡率、Cleaved-caspase3蛋白表达、G0~G1期细胞比例升高,差异均有统计学意义(P < 0.05);相对于pcDNA组,pcDNA-UCA1组AC16细胞存活率、Ki67蛋白表达、S期细胞比例升高,细胞凋亡率、Cleaved-caspase3蛋白表达、G0~G1期细胞比例降低,差异均有统计学意义(P < 0.05)(见图 1、2和表 2)。

图 1 UCA1对缺氧处理的AC16凋亡率
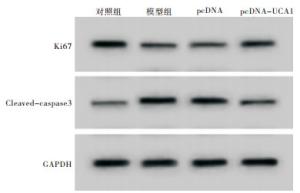
图 2 UCA1对Ki67、Cleaved-caspase3蛋白表达的影响
分组 n 存活率/% 凋亡率/% G0~G1 S G2~M Ki67 Cleaved-caspase3 对照组 3 95.88±5.11 6.73±0.39 33.59±1.16 33.70±1.20 32.71±1.11 0.89±0.05 0.13±0.01 模型组 3 44.94±1.47* 24.40±0.91* 49.10±1.10* 18.41±0.94* 32.49±0.91 0.23±0.01* 0.79±0.05* pcDNA 3 44.45±1.16 24.34±0.91 49.12±1.05 18.51±0.90 32.37±0.93 0.23±0.02 0.80±0.04 pcDNA-UCA1 3 83.25±2.15#△ 12.16±0.50#△ 37.29±0.94#△ 30.38±0.79#△ 32.33±0.70 0.71±0.04#△ 0.24±0.02#△ F — 244.52 461.55 170.61 202.18 0.10 296.61 328.87 P — < 0.01 < 0.01 < 0.01 < 0.01 >0.05 < 0.01 < 0.01 MS组内 — 8.560 0.515 1.135 0.939 0.854 0.001 0.001 q检验:与对照组比较*P < 0.05;与模型组比较#P < 0.05;与pcDNA组比较△P < 0.05 表 2 UCA1对缺氧处理的AC16细胞增殖凋亡的影响(x±s)
-
靶基因预测数据库在线分析显示,UCA1与miR-503之间存在特异性结合位点(见图 3)。双荧光素酶报告实验显示,与转染miR-NC比较,转染miR-503 mimic可降低WT-UCA1的相对荧光素酶活性(P < 0.01),而对MUT-UCA1的相对荧光素酶活性影响无统计学意义(P>0.05)(见表 3)。RT-qPCR检测显示,与pcDNA组(0.96±0.10)比较,pcDNA-UCA1组(0.26±0.02)AC16细胞miR-503的表达水平降低(t=11.89,P < 0.01)。
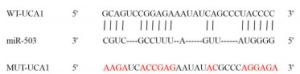
图 3 UCA1和miR-503存在互补序列
分组 n WT-UCA1 MUT-UCA1 miR-NC 3 0.95±0.09 0.90±0.06 miR-503 3 0.38±0.02 0.93±0.06 t — 10.71 0.61 P — < 0.01 >0.05 表 3 双荧光素酶报告实验验证UCA1和miR-503的靶向关系(x±s)
-
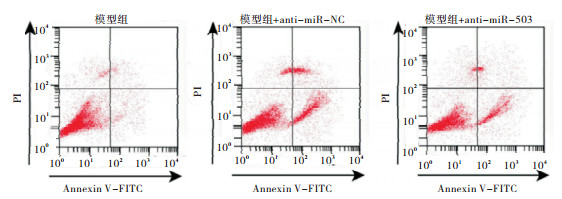
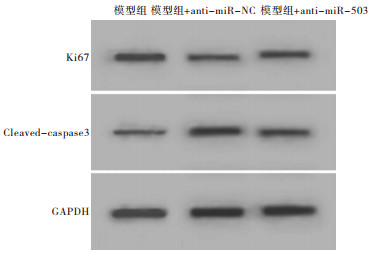
相对于Model组、anti-miR-NC组,anti-miR-503组AC16细胞存活率、Ki67蛋白表达、S期细胞比例显著升高,细胞凋亡率、Cleaved-caspase3蛋白表达、G0~G1期细胞比例显著降低,差异均有统计学意义(P < 0.05)(见图 4、5和表 4)。

图 4 干扰miR-503对缺氧处理的AC16凋亡率

图 5 干扰miR-503对Ki67、Cleaved-caspase3蛋白表达的影响
分组 n 存活率/% 凋亡率/% G0~G1 S G2~M Ki67 Cleaved-caspase3 模型组 3 44.58±3.25 24.42±0.66 49.61±1.17 18.41±1.05 31.98±1.18 0.25±0.02 0.82±0.05 模型组+anti-miR-NC 3 44.79±0.10 24.38±0.77 49.24±0.98 18.38±0.89 32.38±0.94 0.23±0.02 0.78±0.05 Model+anti-miR-503 3 72.97±1.59△ 15.55±0.52△ 40.44±0.79△ 28.12±0.64△ 32.44±0.77 0.56±0.02△ 0.34±0.03△ F — 18.22 180.90 82.18 123.14 0.20 256.75 108.20 P — < 0.01 < 0.01 < 0.01 < 0.01 >0.05 < 0.01 < 0.01 MS组内 — 4.367 0.433 0.985 0.768 0.956 0.000 0.002 q检验:与anti-miR-NC组比较△P < 0.05 表 4 干扰miR-503对缺氧处理的AC16增殖凋亡的影响(x±s)
-
lncRNA作为心血管疾病进展的重要调节剂,与AMI、缺血性心脏病等缺氧诱导的心肌细胞损伤性疾病有关[8]。探讨lncRNA UCA1在缺氧诱导的心肌细胞损伤中的作用,有望为缺氧引起的AMI防治提供有效策略。
研究[9]显示,血浆UCA1较高的慢性心力衰竭(CHF)病人的生存率较低,UCA1低表达对CHF病人生存具有预测作用。UCA1通过与miR-184竞争性结合增强沉默同源盒A9表达从而促进了心脏肥大进程[10]。UCA1通过与miR-206相互作用加剧氧化低密度脂蛋白诱导的巨噬细胞氧化应激和细胞凋亡,对动脉粥样硬化进展具有潜在促进作用[11]。本研究探讨UCA1在AMI中作用发现,缺氧可诱导AC16细胞凋亡和G0~G1期阻滞,抑制细胞存活,诱导AC16细胞损伤。缺氧诱导后AC16细胞中UCA1表达降低,提示UCA1低表达可能与缺氧诱导的细胞损伤有关。转染pcDNA-UCA1上调UCA1表达后,缺氧诱导的AC16细胞凋亡率、G0~G1期细胞比例显著降低,细胞存活、S期细胞比例显著升高。Ki67是一种仅表达增殖期细胞的核内蛋白,与细胞增殖密切相关,是评价细胞增殖能力的常用指标[12]。Cleaved-caspase3为细胞凋亡发生的最终执行因子,在多种凋亡信号刺激下,caspase3裂解为Cleaved-caspase3,促进细胞凋亡[13]。本研究显示,上调UCA1表达可降低缺氧诱导的AC16细胞中Cleaved-caspase3表达水平,提高Ki67表达水平,与功能实验结果一致,说明上调UCA1能够减轻缺氧诱导的AC16细胞损伤。
miR-503是心脏功能和心血管疾病的重要表观遗传调控因子。在患有糖尿病的急性缺血性中风的病人中miR-503明显表达增加[14]。miR-503通过靶向Apelin-13促进结缔组织生长因子和转化生长因子表达,促进胶原蛋白产生,进而促进血管紧张素Ⅱ诱导的心脏纤维化[15]。miR-503低表达还与炎症介导的血管生成有关[16]。此外,lncRNA肺腺癌转移相关转录本1(MALAT1)可能通过抑制miR-503表达抑制内皮细胞黏附和促炎因子分泌,对动脉粥样硬化形成具有抑制作用[17]。本研究通过双荧光素酶报告基因证实,UCA1对miR-503具有靶向调控作用,上调UCA1可降低miR-503的表达水平。此外,缺氧诱导后AC16细胞中miR-503表达升高,转染anti-miR-503下调miR-503表达可提高缺氧条件下AC16细胞存活率、S期细胞比例、Ki67表达水平,降低Cleaved-caspase3表达水平,减轻缺氧诱导的细胞凋亡和G0~G1期阻滞,改善缺氧诱导的AC16细胞损伤,与上调UCA1表达对缺氧诱导AC16细胞损伤的保护作用一致。以上研究提示UCA1可能靶向miR-503保护缺氧诱导的AC16细胞损伤。
综上所述,lncRNA UCA1可能通过靶向下调miR-503表达保护心肌细胞免受缺氧诱导的损伤,提示UCA1是防治缺氧引起的AMI的重要靶点。
lncRNA UCA1抑制miR-503减轻缺氧诱导的心肌细胞损伤机制研究
lncRNA UCA1 alleviates hypoxia-induced cardiomyocyte injury by targeting miR-503
-
摘要:
目的探讨长链非编码RNA(lncRNA)尿路上皮癌相关1(UCA1)靶向miR-503对缺氧条件下心肌细胞增殖、凋亡的影响。 方法构建体外心肌细胞AC16缺氧模型。实时荧光定量PCR(RT-qPCR)检测UCA1和miR-503表达。细胞计数试剂盒(CCK-8)检测细胞活力;流式细胞术检测细胞凋亡和周期分布。将UCA1过表达载体、miR-503抑制物分别转染AC16,检测上调UCA1或下调miR-503对缺氧条件下AC16细胞增殖和凋亡的影响。双荧光素酶报告基因实验和RT-qPCR确定UCA1对miR-503的靶向作用。 结果缺氧处理后AC16细胞存活率、S期细胞比例降低,凋亡率、G0~G1期细胞比例升高,差异均有统计学意义(P < 0.05)。上调UCA1或下调miR-503表达后,缺氧处理的AC16细胞存活率、S期细胞比例升高,凋亡率、G0~G1期细胞比例降低,差异均有统计学意义(P < 0.05)。miR-503是UCA1的靶基因,UCA1负性调控miR-503表达。 结论lncRNA UCA1能够显著提高缺氧条件下心肌细胞存活率,抑制缺氧诱导的心肌细胞凋亡,其机制可能与靶向下调miR-503表达有关。 Abstract:ObjectiveTo investigate the effect of long-chain non-coding RNA(lncRNA) urothelial carcinoma is associated 1(UCA1) targeting miR-503 on proliferation and apoptosis of cardiomyocyte with hypoxia treatment. MethodsThe cardiomyocyte AC16 hypoxia model was established in vitro.The expression of UCA1 and miR-503 were detected by real-time quantitative PCR (RT-qPCR).The cell viability was measured by CCK-8.Apoptosis and cycle distribution were detected by flow cytometry.The UCA1 overexpression vector and miR-503 inhibitor were transfected into AC16 cells, respectively.And the effects of up-regulating UCA1 or down-regulating miR-503 on the proliferation and apoptosis of AC16 cells treated with hypoxic were detected.Dual luciferase reporter gene assay and RT-qPCR confirmed the targeting effect of UCA1 on miR-503. ResultsAfter treated with hypoxia, the survival rate of AC16 cells and the proportion of S-phase cells were significantly decreased, and the apoptosis rate and the proportion of G0-G1 phase cells were significantly increased (P < 0.05).After up-regulating UCA1 expression or down-regulating miR-503 expression, the survival rate and the proportion of S-phase in AC16 cells treated with hypoxia were significantly increased, while the apoptosis rate and the proportion of G0-G1 phase cells were significantly decreased (P < 0.05).miR-503 was the target gene of UCA1, and UCA1 negatively regulated the expression of miR-503. ConclusionslncRNA UCA1 can significantly improve the survival rate and inhibit the apoptosis in hypoxia-induced cardiomyocyte, which may be attributed to the targeted down-regulation of miR-503 expression. -
表 1 UCA1、miR-503在缺氧处理的AC16细胞中的表达(x±s)
分组 n UCA1 miR-503 对照组 3 0.99±0.09 0.96±0.10 模型组 3 0.12±0.02 2.67±0.12 t — 16.34 18.96 P — < 0.01 < 0.01  下载: 导出CSV
下载: 导出CSV
表 2 UCA1对缺氧处理的AC16细胞增殖凋亡的影响(x±s)
分组 n 存活率/% 凋亡率/% G0~G1 S G2~M Ki67 Cleaved-caspase3 对照组 3 95.88±5.11 6.73±0.39 33.59±1.16 33.70±1.20 32.71±1.11 0.89±0.05 0.13±0.01 模型组 3 44.94±1.47* 24.40±0.91* 49.10±1.10* 18.41±0.94* 32.49±0.91 0.23±0.01* 0.79±0.05* pcDNA 3 44.45±1.16 24.34±0.91 49.12±1.05 18.51±0.90 32.37±0.93 0.23±0.02 0.80±0.04 pcDNA-UCA1 3 83.25±2.15#△ 12.16±0.50#△ 37.29±0.94#△ 30.38±0.79#△ 32.33±0.70 0.71±0.04#△ 0.24±0.02#△ F — 244.52 461.55 170.61 202.18 0.10 296.61 328.87 P — < 0.01 < 0.01 < 0.01 < 0.01 >0.05 < 0.01 < 0.01 MS组内 — 8.560 0.515 1.135 0.939 0.854 0.001 0.001 q检验:与对照组比较*P < 0.05;与模型组比较#P < 0.05;与pcDNA组比较△P < 0.05
下载: 导出CSV
表 3 双荧光素酶报告实验验证UCA1和miR-503的靶向关系(x±s)
分组 n WT-UCA1 MUT-UCA1 miR-NC 3 0.95±0.09 0.90±0.06 miR-503 3 0.38±0.02 0.93±0.06 t — 10.71 0.61 P — < 0.01 >0.05
下载: 导出CSV
表 4 干扰miR-503对缺氧处理的AC16增殖凋亡的影响(x±s)
分组 n 存活率/% 凋亡率/% G0~G1 S G2~M Ki67 Cleaved-caspase3 模型组 3 44.58±3.25 24.42±0.66 49.61±1.17 18.41±1.05 31.98±1.18 0.25±0.02 0.82±0.05 模型组+anti-miR-NC 3 44.79±0.10 24.38±0.77 49.24±0.98 18.38±0.89 32.38±0.94 0.23±0.02 0.78±0.05 Model+anti-miR-503 3 72.97±1.59△ 15.55±0.52△ 40.44±0.79△ 28.12±0.64△ 32.44±0.77 0.56±0.02△ 0.34±0.03△ F — 18.22 180.90 82.18 123.14 0.20 256.75 108.20 P — < 0.01 < 0.01 < 0.01 < 0.01 >0.05 < 0.01 < 0.01 MS组内 — 4.367 0.433 0.985 0.768 0.956 0.000 0.002 q检验:与anti-miR-NC组比较△P < 0.05
下载: 导出CSV
-
[1] ANDERSON JL, MORROW DA. Acute Myocardial Infarction[J]. N Engl J Med, 2017, 376(21): 2053. doi: 10.1056/NEJMra1606915 [2] FRANGOGIANNIS NG. Pathophysiology of Myocardial Infarction[J]. Compr Physiol, 2015, 5(4): 1841. [3] POLLER W, DIMMELER S, HEYMANS S, et al. Non-coding RNAs in cardiovascular diseases: diagnostic and therapeutic perspectives[J]. Eur Heart J, 2018, 39(29): 2704. doi: 10.1093/eurheartj/ehx165 [4] LIU Z, YAN Y, CAO S, et al. Long non-coding RNA SNHG14 contributes to gastric cancer development through targeting miR-145/SOX9 axis[J]. J Cell Biochem, 2018, 119(8): 6905. doi: 10.1002/jcb.26889 [5] 张滨. 长链非编码RNA(UCA1)在急性心肌梗死患者血浆中变化水平的研究[D]. 长春: 吉林大学, 2015. [6] 陈琴, 黄铭涵, 张在保, 等. microRNA-503在糖尿病大鼠心肌梗死后血管新生过程中的变化[J]. 中国动脉硬化杂志, 2015, 23(3): 261. [7] 宋舒璇. miR-29b1在缺氧诱导心肌细胞损伤中的作用机制研究[D]. 广州: 广州医科大学, 2018. [8] SALLAM T, SANDHU J, TONTONOZ P. Long noncoding RNA discovery in cardiovascular disease: decoding form to function[J]. Circ Res, 2018, 122(1): 155. doi: 10.1161/CIRCRESAHA.117.311802 [9] YU X, ZOU T, ZOU L, et al. Plasma long noncoding RNA urothelial carcinoma associated 1 predicts poor prognosis in chronic heart failure patients[J]. Med Sci Monit, 2017, 23(1): 2226. [10] ZHOU G, LI C, FENG J, et al. lncRNA UCA1 is a novel regulator in cardiomyocyte hypertrophy through targeting the miR-184/HOXA9 Axis[J]. Cardiorenal Med, 2018, 8(2): 130. doi: 10.1159/000487204 [11] HU X, MA R, FU W, et al. LncRNA UCA1 sponges miR-206 to exacerbate oxidative stress and apoptosis induced by ox-LDL in human macrophages[J]. J Cell Physiol, 2019, 234(8): 14154. doi: 10.1002/jcp.28109 [12] 李冰, 周平, 靳义. 吉马酮对人非小细胞肺癌NCI-H1770细胞增殖、凋亡、侵袭和迁移的调节作用[J]. 中国免疫学杂志, 2019, 35(7): 819. doi: 10.3969/j.issn.1000-484X.2019.07.010 [13] 张胜强, 张洪艳, 齐宝林, 等. 三叶青黄酮诱导肺癌SPC-A-1细胞凋亡与cleaved-caspase-3表达的关系[J]. 中国医院药学杂志, 2017, 37(12): 1139. [14] SHEIKHBAHAEI S, MANIZHEH D, MOHAMMAD S, et al. Can MiR-503 be used as a marker in diabetic patients with ischemic stroke?[J]. BMC Endocr Disord, 2019, 19(1): 42. doi: 10.1186/s12902-019-0371-6 [15] ZHOU Y, DENG L, ZHAO D, et al. MicroRNA-503 promotes angiotensin Ⅱ-induced cardiac fibrosis by targeting Apelin-13[J]. J Cell Mol Med, 2016, 20(3): 495. doi: 10.1111/jcmm.12754 [16] LEE A, PAPANGELI I, PARK Y, et al. A PPARγ-dependent miR-424/503-CD40 axis regulates inflammation mediated angiogenesis[J]. Sci Rep, 2017, 7(1): 2528. doi: 10.1038/s41598-017-02852-4 [17] CREMER S, MICHALIK KM, FISCHER A, et al. Hematopoietic deficiency of the long noncoding RNA MALAT1 promotes atherosclerosis and plaque inflammation[J]. Circulation, 2019, 139(10): 1320. doi: 10.1161/CIRCULATIONAHA.117.029015 -

 点击查看大图
点击查看大图

图(5)表(4)
计量
- 文章访问数: 4000
- HTML全文浏览量: 2829
- PDF下载量: 8
- 被引次数: 0
