




-
Alport综合征(alport syndrome,AS)又称遗传性进行性肾炎,临床特点是以血尿、蛋白尿及进行性肾功能减退为主,部分病人可合并出现感音神经性耳聋、眼部异常、食管平滑肌瘤等肾外表现,是危及儿童及青少年生命健康的重要疾病之一[1-3]。随着精准医学的发展及各级医疗机构诊疗水平的提高, 其发病率呈逐年上升趋势。根据2008年北美儿童肾脏实验及合作性研究中心的数据显示,AS患儿肾移植占同期儿童肾移植的2.3%, 占同期儿童血液透析的2.3%, 该病活产儿发病率为1:5000[4]。AS为编码Ⅳ型胶原α3、α4、α5链的基因突变所致,其中α3(Ⅳ)、α4(Ⅳ)基因突变所致为常染色体显性或隐性遗传,α5(Ⅳ)基因突变所致为X连锁显性遗传,至今为止已有超出700个基因突变被发现[5]。本研究采用高通量第2代测序技术对1例AS患儿进行检测,结果发现一个新的α5链位点移码突变,为国内外首次报道。
HTML
-
患儿,女,3岁11个月,因“发现血尿、蛋白尿2年,水肿3d,咳嗽1d”于2018年12月就诊于安徽省儿童医院肾内科。患儿入院2年前(2016年12月)曾因“肺炎”在当地医院住院治疗时尿检发现隐血阳性,入院5个月前当地复诊复查尿检提示尿蛋白2+,隐血3+,血清白蛋白未见异常,24h尿蛋白未见异常,当地医院诊断:血尿待查,IgA肾病可能,入院2个月前当地复查, 尿蛋白弱阳性,红细胞计数849.9~1274.7/μL;红细胞形态:非均一性红细胞;予以“黄葵胶囊”对症处理。患儿入院3d前出现咳嗽及双眼睑水肿伴尿少,尿色呈浓茶色。病程中无皮疹,无关节肿痛。
入院查体:体温36.1 ℃, 脉搏110次/分,呼吸30次/分,体质量19.9 kg,血压108/60 mmHg, 双眼睑水肿,咽部充血,双肺呼吸音粗糙,可闻及中粗湿啰音,心腹查体无特殊。诊断:肾病综合征(肾炎型),给予积极抗感染治疗后肉眼血尿缓解。建议基因检查排除AS,送检第2代高通量测序(北京智因东方转化医学研究中心,北京全谱医学检验实验室)。患儿系母亲第1胎第1产,足月顺产出生,无窒息抢救史,胎盘及羊水无特殊,生长发育史无异常。父母非近期婚配,均体健。否认家族遗传病史及传染病接触史。我院实验室检查,尿常规:外观微混,颜色淡红,pH5.5,尿蛋白3+,红细胞5162/μL、929/HP、非均一性小细胞,24 h尿蛋白2.11g/L;尿敏感肾功能六项:尿微量白蛋白1 582.40 mg/L,尿免疫球蛋白IgG 30.2 mg/L, N-乙酰-β-D-葡萄糖苷酶13.50 U/L, 4-尿转铁蛋白110.15 mg/L;生化:白蛋白25.9 g/L, 尿素6.8 mmol/L,血肌酐38.7 μmol/L;血脂分析:总胆固醇5.81 mmol/L,钙2.15 mmol/L。肝功能正常,ASO 3.2IU/mL, 补体C3 1.6 g/L, 补体C4 0.14 g/L, 免疫球蛋白M 2.32 g/L, 出凝血时间正常,清洁中段尿培养未见细菌,双肾B超示:右肾7.0 cm×3.5 cm,左肾6.9 cm×3.4 cm, 双肾弥漫性病变。左肾静脉彩超未见异常,纯音测听听力无障碍,眼裂隙灯检查及视力检查未见异常,患儿父母及近三代尿常规未见异常。
入院后积极控制呼吸道感染予以头孢西丁抗感染,雾化化痰;添加缬沙坦27 mg每天1次,降蛋白对症治疗。患儿发病年龄小(1年11个月),肾脏出现损害时间长(2年),考虑遗传代谢病,需行肾活检和基因检测。与家长沟通后于2019年1月9日肾活检,结果提示基底膜厚薄不一,致密层增厚,部分呈撕裂状和蛛网状,建议进一步完善基因检测。2019年1月15日复查尿蛋白转阴,尿敏感肾功能六项:尿微量白蛋白30.39 mg/L,尿免疫球蛋白IgG 2.4 mg/L, N-乙酰-β-D-葡萄糖苷酶10.16 U/L, 4-尿转铁蛋白2.75 mg/L,初期未添加激素等免疫抑制剂,临床症状及辅检较前明显好转,目前予以缬沙坦27 mg每天1次及百令胶囊对症治疗。4个月后复诊,尿蛋白未见异常,肾功能正常,尿微量白蛋白30.00 mg/L,轻度升高。
-
采用B超定位下经皮肾活检术,活检肾组织按常规方法进行光镜、免疫荧光和电镜检查,加做Ⅳ型胶原的检测。
-
在患儿家长知情同意的前提下,采集病人及父母全血2 mL(EDTA抗凝血)进行基因分析。基因组DNA提取:取病人新鲜外周血,使用BloodGen Midi Kit(CWBIO, China)提取病人全基因组DNA,操作按照试剂盒说明书进行。
-
采用IDT公司xGen® Exome Research Panel v1.0捕获芯片。Cavoris仪打断至200 bp左右,片段化DNA在Klenow Fragment, T4 DNA polymerase和T4PNK的作用下进行末端补平修复。在聚合酶体系下,使上一步得到的修复产物在3′末端加上A碱基,为下一步连接做好准备。配置T4DNA连接酶反应体系,在Thermo mixer中适温反应一定时间使adapter和加“A”产物连接。连接产物经4~6轮LM-PCR扩增。文库与探针混于杂交体系中65 ℃、60~68 h杂交。使用链霉素磁珠,与杂交样本孵育后洗脱液洗脱。洗脱产物经10轮LM-PCR扩增。Illumina hiseq xten平台标准化上机测序操作流程,测序得到图像原始数据,Illumina官方basecall分析软件BclToFastq得到原始数据(raw data)。去接头污染,去低质量的数据,数据与参考序列比对统计(比对软件BWA),参考基因组:hg19基因组。SNP检测及注释:分析软件samtools。Indel检测及注释:分析软件pindel。变异假阳性过滤:根据测序深度,变异质量,对检测得到的SNP、Indel进行过滤筛选,得到高质量可靠的变异。变异注释:SNP和Indel根据在基因上的位置,分析得到氨基酸变化影响、剪切影响、UTR、内含子变异影响等。使用SIFT利用基于同源比对,蛋白结构的保守性等的算法,预测筛选出的变异对蛋白质的影响。对剪切位点附近的变异,做剪切危害性预测。
-
根据COL4A5基因验证位点序列设计引物,应用PCR方法进行扩增。PCR反应条件为:95 ℃预变性5 min,95 ℃变性30 s,65 ℃退火30 s, 72 ℃链延伸30 s,扩增30个循环。最后72 ℃补充延伸10 min。PCR的体系均为50 μL。用ABI 3730XL测序仪测序,测序引物采用原PCR引物。基因序列分析采用DNASTAR软件进行序列分析和比对,mRNA比对模板为NM_033380。对点变异携带者病人及父母样本进行Sanger测序。
1.1. 临床资料
1.2. 病理研究方法
1.3. DNA测序研究方法
1.3.1. 基因组DNA提取
1.3.2. 全外显子组测序
1.3.3. Sanger测序验证
-
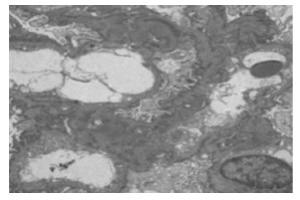
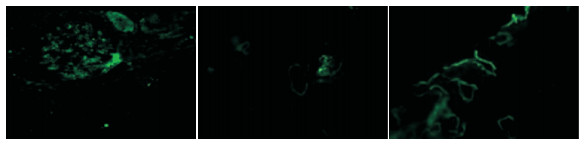
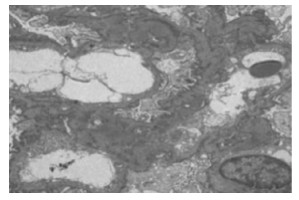
综合光镜、免疫及电镜结果回报:肾小球轻微病变伴个别新月体形成,C3系膜区沉积,可见19个肾小球,将荧光备片经PASM染色,可见12个肾小球,未见肾小球球性及节段性硬化。肾小球系膜细胞和基质轻微增生,无明显局灶节段性加重及内皮细胞增生,系膜区少许嗜复红蛋白沉积,毛细血管襻开放,荧光备片中1个细胞性新月体形成。肾小管上皮细胞空泡及颗粒变性,少数肾小管管腔扩张,刷毛缘消失,个别肾小管萎缩,肾间质轻度水肿,小灶状淋巴细胞及少许嗜酸性粒细胞浸润,小动脉管壁无明显病变(见图 1)。免疫荧光:C3+球性系膜区呈颗粒状沉积;a3、a5肾小管基底膜节段表达(见图 2)。超微结果:基底膜厚薄不一,基底膜致密层增厚,部分呈撕裂状和蛛网状,足突弥漫融合,少数系膜区可见少量电子致密物沉积(见图 3)。


-
全外显子组测序结果显示病人COL4A5基因49号外显子存在c.4 518_c.4 519 insC(p.Gln1 507Profs*14)的杂合变异,通过Sanger测序证实;该变异为第48号外显子上4 518和4 519位碱基中间插入了一个胞嘧啶,导致蛋白在第1 507位谷氨酰胺变异为脯氨酸后再编码14个密码子即终止编码,造成合成的蛋白出现截短,破坏了C末端的非胶原结构域NC1(见图 4)。另该变异在dbSNP、Hapmap、千人基因组(1000 Genomes)、ExAC、gnomAD等数据库分布频率均为0。根据《ACMG遗传变异分类标准与指南》[6]的变异致病证据分级,病人携带的该变异满足PV1+PS1+PM2的致病证据,达到致病性变异(Pathogenic)级别。同时也未检测到病人其他肾炎相关基因的可疑致病变异。病人父母均不携带该变异,在该变异位置均为野生型,验证结果与目标序列捕获测序一致。

2.1. 肾活检结果
2.2. 全外显子组测序及Sanger测序

-
AS是目前导致儿童终末期肾病(ESRD)的重要疾病之一[3]。由于遗传性疾病现阶段进展至ESRD的治疗方法有限,过程无法逆转,因此儿科医生应高度关注,指导先证者家庭优生优育,降低患病儿童出生率,大大减轻家庭的负担。
本例全外显子组测序结果显示病人COL4A5基因49号外显子存在c.4518_c.4519 insC(p.Gln1507Profs*14)的杂合变异,通过Sanger测序证实。变异造成NC结构域的缺陷可能会引起Ⅳ型胶原蛋白质网络异常,致使基底膜不能分离和支撑许多组织中的细胞行为而发病,详细的致病机制需要进一步的体外实验来探究。WANG等[7]发现COL4A5基因移码变异导致的X连锁AS的一个大家庭,且该家庭的任何病人都没有眼睛或听力异常,仅仅有肾脏病变,没有肾外病变,与本例病人情况类似。本例病人父母该变异均为野生型,也符合孟德尔遗传规律。经检索Human Gene Mutation Database、Clinical Genome Resource、Pubmed和万方、知网等数据库,均未收录COL4A5基因的该变异,为未见报道的新的变异,该先证者基因位点为首次报道新位点,丰富了目前COL4A5基因移码突变谱及表型信息。
COL4A5基因突变位点与临床表现严重程度分为3类:严重型(S型)、中度-重度严重型(M-S型)和中度型(M型)。S型青少年期起病,年龄在20岁以下,80%患儿伴有听力异常,40%患儿伴有视觉异常,为大片段重组或者提前终止翻译或者移框突变或者N端序列突变等。M-S型在26岁前进展至ESRD,肾外系统受累较少,主要基因异常在21~47号外显子区发生甘氨酸替代突变,非甘氨酸错义突变和框内或受体剪切位点突变等。M型则在1~20号外显子区,在30岁后进展至ESRD,70%病人伴有听力丧失,最多30%病人伴有眼部病变。结合本例先证者移码突变位于第48号外显子上,患儿发病年龄早(1岁11个月),临床表现肾脏损害严重(肾病综合征合并肉眼血尿),由于患儿年幼(3岁11月),目前肾外受累不明显,考虑为S型。
有学者[8]建议对每一个AS家系均进行遗传型诊断,以利于对先证者进行预后评估和先证者及其家系进行遗传咨询。AS病人子代患病的概率是有规律可循的:女性病人与正常男性婚配,其子女各1/2发病;男性病人与正常女性婚配,女儿都是病人,儿子正常。常染色体隐性遗传性者:病人双亲无病;同胞中1/4发病,男女机会相等,病人发生在同一代;近亲婚配者发病风险大大增高。AS病人大多数于青壮年时期发展至ESRD,需要透析和肾移植治疗。因此对于AS这样一种预后差的遗传性肾脏病,早期、正确的诊断十分重要。
 DownLoad:
DownLoad: