



-
急性冠状动脉综合征(ACS)是临床常见的危重疾病,是在冠状动脉粥样硬化斑块固定存在的基础上,继发斑块侵袭与破裂,形成完全或不完全闭塞性血栓导致的一组临床综合征[1]。其包括:不稳定型心绞痛(UAP)、非ST段抬高型心肌梗死和ST段抬高型心肌梗死。据报道,2015年我国农村急性心肌梗死(AMI)死亡率达到70.09/10万人,城市死亡率为56.38/10万,住院总费用高达152.40亿元[2],心血管疾病发病率及死亡率增长势头未减,已成为危害我国公民健康的主要因素,故掌握冠心病的发病机制、寻找更精准的病情评估指标具有十分重要的意义,而无创、易获取的血液学指标成为重点筛查对象。
程序性坏死是细胞受到精密的程序调控而发生的坏死性死亡,是非caspase依赖的细胞死亡通路,相关研究已证实其参与冠状动脉斑块形成、心肌缺血再灌注损伤、心肌细胞坏死及纤维化心室重塑等多个病理过程[3-5]。其中刺激受体相互作用蛋白1(RIP1)-RIP3-混合谱系激酶结构域样假激酶(MLKL)是研究较为透彻的经典的通路。KARUNAKARAN等[6]发现MLKL在动脉粥样硬化病人的斑块核内是高度活跃的,说明程序性坏死可能是导致冠状动脉易损性的始动因子之一,可作为诊断及治疗靶点。但目前仍缺乏大样本的临床试验证明MLKL及ACS之间的关系。本研究选取对照组、UAP及AMI组,对比观察RIP1、RIP3和MLKL的组间表达差异,探讨RIP1-RIP3-MLKL通路和ACS疾病进程之间的关系。现作报道。
HTML
-
选取2018年1月至2019年6月经门诊或急诊收住入院的初诊ACS的病人60例,其中UAP病人31例,AMI病人20例,以上病人入院后均经冠状动脉造影证实并成功实施冠状动脉支架植入术。并选取同期健康体检者19名为对照组,均无冠心病病史及心绞痛相关症状。UAP诊断参考2011年美国心脏病学学会基金会/美国心脏协会(ACCF/AHA)的诊断标准[7]。AMI诊断标准参考2012ESC的诊断标准[8]。排除标准包括:(1)有明确的恶性肿瘤病史;(2)严重的肝肾功能不全;(3)有明确的血液系统疾病;(4)有明确的免疫性疾病等炎性疾病;(5)有明确的甲状腺功能亢进等甲状腺疾病;(6)有其他引起胸痛的原因如肺栓塞、心肌病、主动脉瘤、心脏瓣膜病及充血性心力衰竭等;(7)上呼吸道、肝脏、肾脏等感染性疾病;(8)糖尿病等慢性代谢性疾病。研究经蚌埠医学院第一附属医院伦理委员会批准,入选对象均签署了知情同意书。
-
清晨或急诊冠状动脉介入术前采集受试者空腹外周静脉血共约12 mL,分为10 mL及2 mL两份,肝素抗凝;参照朱建等[9]方法分离外周血单个核细胞,流式细胞仪检测,计算CD14阳性率;另一份2 mL外周静脉血经2 500 r/min离心5 min后收集上层血浆,-80 ℃冻存备用。
-
严格按照ELISA试剂盒说明书的步骤进行操作,待检测样品及标准品均为复孔上样,最后在酶标仪上读取450 nm处吸光度值,求平均值并建立标准曲线,按照得出的公式计算出对应样品血浆中IL-1、IL-18及MLKL的含量。
-
采用Trizol法提取外周血单核细胞(PBMCs)总RNA,提取后的RNA A260/A280均在1.8~2.0,取1 μg RNA进行逆转录,反应条件为:42 ℃ 1 h,70 ℃ 5 min,获得的cDNA按照试剂盒(TaKaRa)说明进行qRT-PCR反应。反应体系为:SYBR Green PCR Master Mix(2×)10 μL,cDNA模板2 μL,上、下游引物(10 μmol/L)各0.5 μL,dd H2O 7 μL,反应总体积为20 μL。反应条件为:95 ℃预变性3 min,循环参数95 ℃变性30 s,55 ℃退火30 s,72 ℃延伸30 s,共40个循环,72 ℃ 5 min终止反应。引物由上海生物工程有限公司合成,以人β-actin为内参。MLKL引物序列为:上游引物5′-CAG AAA CAT CAG CAG CTC CA-3′,下游引物5′-ATG ATT TCC CGC AAC AAC TC-3′。β-actin引物序列为5’-CCT TCC TGG GCA TGG AGT CCT G-3′(上游),5′-GGA GCA ATG ATC TTG ATC TTC-3′(下游),产物大小为202 bp。qRT-PCR反应结束后取扩增产物进行2 %琼脂糖凝胶电泳目的条带,扩增结果采用2-△△Ct法分析。
-
提取PBMCs总蛋白,采用BCA蛋白定量试剂盒测定蛋白含量,每孔取20 μg蛋白95 ℃变性后上样,十二烷基硫酸钠-聚丙烯酰胺凝胶100 V、200 mA电泳分离样品蛋白,转印至PVDF膜,用脱脂奶粉室温封闭1 h,加入MLKL、RIP1和RIP3一抗(1∶1 000,Sigma),置摇床4 ℃孵育过夜,经PBST洗膜3次后加入HRP标记二抗(1∶5 000,Amersham),37 ℃孵育,室温振摇2 h。洗膜后取5 mL BCIP/NBT显色液(Invitrogen)按照显色试剂说明书进行显色。用Quality图像分析系统对蛋白条带进行灰度分析,β-actin的表达水平作为内参。
-
采用方差分析、q检验和χ2检验。
1.1. 一般资料
1.2. 样本制备
1.3. ELISA法检测外周血浆中白细胞介素(IL)-1、IL-18及MLKL的含量
1.4. qRT-PCR提取核RNA
1.5. Western blotting
1.6. 统计学方法
-
3组间年龄、高血压、糖尿病、白细胞(WBC)、红细胞(RBC)、血小板(PLT)、三酰甘油、高密度脂蛋白(HDL)、低密度脂蛋白(LDL)、载脂蛋白A、丙氨酸氨基转移酶、总胆红素(TBil)、总蛋白差异均无统计学意义(P>0.05);而AMI组中血清总胆固醇(TC)、天冬氨酸氨基转移酶和血肌酐(Cr)水平明显高于UAP组和对照组(P < 0.05)(见表 1)。
分组 n 年龄/岁 高血压 糖尿病 WBC/
(×109/L)RBC/
(×1012/L)PLT/
(×109/L)TC/
(mmol/L)三酰甘油/
(mmol/L)对照组 19 59.74±7.56 18 3 6.35±2.78 4.28±0.76 235.37±116.24 3.93±1.30 1.52±1.23 UAP组 31 63.87±9.59 12 7 6.62±1.65 4.35±0.44 218.45±68.70 3.94±1.05 1.71±0.83 AMI组 20 64.75±7.78 9 3 7.62±2.97 4.18±0.69 266.55±78.50 5.04±2.12*# 1.76±0.74 F — 1.95 1.01△ 0.66△ 1.57 0.47 1.88 3.92 0.37 P — >0.05 >0.05 >0.05 >0.05 >0.05 >0.05 < 0.05 >0.05 MS组内 — 73.699 — — 5.797 0.377 7 490.817 2.222 0.870 分组 n HDL/
(mmol/L)LDL/
(mmol/L)天冬氨酸氨基
转移酶/(IU/L)丙氨酸氨基
转移酶/(IU/L)TBil/
(μmol/L)总蛋白/
(g/L)Cr/
(mg/L)对照组 19 1.06±0.26 2.12±0.71 20.42±3.52 21.70±11.56 9.83±3.81 65.71±5.61 59.00±14.14 UAP组 31 0.93±0.17 2.21±0.69 20.97±6.90 19.16±10.60 10.32±4.39 65.87±4.97 65.52±10.72 AMI组 20 0.94±0.27 2.65±1.02 32.35±18.97*# 19.79±10.50 10.10±5.60 66.29±7.29 79.78±42.88* F — 2.14 2.57 7.59 0.33 0.07 0.05 3.59 P — >0.05 >0.05 < 0.01 >0.05 >0.05 >0.05 < 0.05 MS组内 — 0.052 0.644 126.697 117.477 21.426 34.586 626.592 △示χ2值: q检验:与对照组比较*P < 0.05;与UAP组比较#P < 0.05 -
3组受试者血浆IL-1、IL-18含量和MLKL含量差异均有统计学意义(P < 0.01)(见表 2)。
分组 n IL-1/(pg/mL) IL-18/(pg/mL) MLKL/(ng/mL) 对照组 19 99.49±21.44 121.35±29.02 517.65±163.97 UAP组 31 126.19±20.79* 136.89±41.42 646.06±166.33* AMI组 20 146.60±17.32*# 168.45±35.13*# 745.03±180.70*# F — 27.03 52.00 8.77 P — < 0.01 < 0.01 < 0.01 MS组内 — 402.097 1 344.412 28 870.42 q检验:与对照组比较*P < 0.05;与UAP组比较#P < 0.05 -
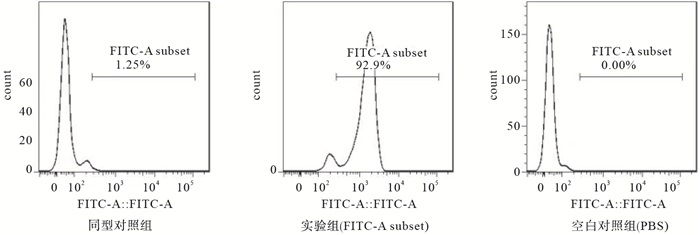
PBMCs经CD14-抗体结合磁珠提纯并经流式鉴定,得到的人PBMCs纯度>90%(见图 1)。

-
qRT-PCR结果显示UAP组MLK mRNA表达量较对照组明显升高(P < 0.05),AMI组MLKL mRNA高于对照组和UAP组(P < 0.05)(见表 3)。
分组 n MLKLmRNA 对照组 19 1.00 UAP组 31 1.17±0.17* AMI组 20 1.56±0.34*# F — 36.726 P — < 0.01 MS组内 — 3.045 q检验:与对照组比较*P < 0.05;与UAP组比较#P < 0.05 -
与对照组比较,UAP组MLKL、RIP1和RIP3的蛋白表达升高(P < 0.05);与UAP组比较AMI组MLKL、RIP1和RIP3的蛋白表达进一步升高(P < 0.05)(见表 4)。
分组 MLKL RIP1 RIP3 对照组 0.84±0.12 0.95±.08 0.82±0.10 UAP组 0.97±0.07* 1.03±.08* 0.93±0.14* AMI组 1.18±0.08*# 1.32±0.10*# 1.22±0.18*# F 72.43 91.67 41.17 P < 0.01 < 0.01 < 0.01 MS组内 0.520 0.541 0.022 q检验:与对照组比较*P < 0.05;与UAP组比较#P < 0.05
2.1. 3组临床资料比较
2.2. 3组外周血浆IL-1、IL-18和MLKL含量比较
2.3. PBMCs的鉴定
2.4. PBMCs中MLKL的mRNA表达
2.5. PBMCs中MLKL、RIP1、RIP3的蛋白表达
-
冠心病的发生发展和环境、行为及遗传因素等息息相关。其中,高胆固醇水平是重要的危险因素,长期脂质代谢紊乱,导致巨噬细胞吞噬脂质形成泡沫细胞,加速斑块形成。炎症同样是动脉粥样硬化的重要驱动因素,而晶体胆固醇作为内源性危险信号,沉积在动脉壁或其他部位是炎症的早期原因[10]。心脑血管疾病中,最常见的是NLRP3炎症反应体,由NLRP3、Asc和caspase 1组成,这种多聚复合物的激活启动下游反应,包括促炎症细胞因子IL-1β和IL-18的成熟[11]。IL-1β和其他IL家族细胞因子是重要的血管和全身炎症介质,同时也在冠心病发生发展扮演重要角色[12]。因此本研究选取IL-1、IL-18作为炎症观察指标,发现UAP病人IL-1、IL-18水平较正常对照组增加,这提示斑块形成过程是长期低水平的促炎状态,随着病程进展,在斑块脱落或堵塞管腔的一瞬间,机体产生促炎风暴。研究亦显示AMI组较UAP组IL-1、IL-18进行性升高。SANDANGER等[13]发现敲除ASC和NLRP 3基因也显示梗死范围明显缩小并保留心功能,因此IL-1、IL-18作为心肌梗死的危险性因素,其下调有助于心功能的恢复。
RIP1-RIP3-MLKL是近年的研究热点通路。既往我们所熟知的细胞因子,如RIP1、Fas相关蛋白结合死亡域(FADD)、TNF受体相关因子2(TRAF 2)、肿瘤坏死因子α(TNF-α)和caspase 8等,均参与凋亡程序中[14]。而当caspase 8的状态被抑制或MLKL的表达水平较高,细胞可发生程序性死亡。其中,RIP1和RIP3是关键激酶,MLKL磷酸化也是死亡的直接执行者,MLKL的苏氨酸357和丝氨酸358结构域被RIP3磷酸化,激活下游反应,诱导程序性坏死程序的发生[15-19]。巨氧化型低密度脂蛋白(OX-LDL)可以在生理状态下通过直接促进巨噬细胞中RIP3和MLKL的表达及活化诱导程序性坏死[6]。然后泡沫细胞在血管壁内发生坏死,增加斑块不稳定性。UAP时期,狭窄的冠状动脉痉挛,或运动后不稳定斑块表面纤维帽破裂,血小板黏附,造成心肌细胞短时间的缺血缺氧,产生痛感,病人因此入院。本研究显示,UAP时期病人PBMCs中RIP1、RIP3和MLKL水平明显上调,PCR及Elisa检测MLKL水平也上调,说明这一时期,MLKL表达增多,缺氧的心肌细胞在TNF-α的介导下,程序性坏死水平激活。
心肌缺血再灌注损伤指的是硬化的冠状动脉部分或完全急性阻塞后一定时间内血液再通时,缺血的心肌虽然血供得以恢复,但其组织损伤有时却呈进行性不可逆加重[20]。OERLEMANS等[21]发现,缺血再灌注损伤后的心肌细胞中RIP1、RIP3和MLKL蛋白含量明显上调,NEC-1,一种特异性程序性坏死抑制剂,能抑制RIP1/3磷酸化和MLKL募集,下调RIP1、RIP3和MLKL,减轻心肌细胞损伤,提高对氧化应激的抵抗力和减少线粒体功能障碍,对心肌缺血/再灌注后短期和长期心功能的恢复都是有益的。有鉴于此,本研究旨在探讨较正常人群比较,ACS病人血浆及PBMCs中程序性坏死通路关键因子的表达变化,为此收集20例心肌梗死病人血标本,发现AMI组病人外周血浆中MLKL进一步升高,RIP1和RIP3的蛋白表达的上调,MLKL的mRNA和蛋白表达上调。也侧面说明长期心肌细胞缺氧时,全面调动体内炎症水平的同时,程序性坏死也是高度激活的。也有报道说明程序性坏死可能参与肌钙蛋白形成过程[22]。我们推测:AMI时,心肌细胞不可逆损伤,炎症因子大幅释放,RIP1、RIP3和MLKL上调,与此同时高表达的程序性坏死,增加细胞膜内外离子浓度梯度,加速心肌细胞膜破裂,加剧体内炎症水平。从正常组至UAP及AMI,随着心肌细胞损伤程度增加,RIP1、RIP3和MLKL表达递增。RIP3除了通过细胞凋亡和炎症外,还通过激活Ca2+-钙调素依赖性蛋白激酶(CaMKII)而不是我们上述的经典的RIP1、RIP3和MLKL来触发心肌细胞坏死[23]。RIP3缺乏或CaMKII抑制可改善缺血再灌注或阿霉素治疗所致的心肌坏死和心力衰竭[24]。
综上所述,本研究发现程序性坏死通路参与ACS病程始终,与ACS病人体内炎症状态密切相关。从RIP1-RIP3-MLKL在不同类型ACS病人中表达的差异,我们推测血浆RIP1、RIP3和MLKL是的ACS危险因素,可作为ACS筛查的替代标志物。血浆和PBMCs中MLKL等水平与心肌细胞损伤程度及其临床表现呈正相关。这表明血浆MLKL有望成为预测ACS疾病的严重程度的指标,为ACS早期发现、早期诊断、早期治疗提供莫大的帮助。
 DownLoad:
DownLoad: